
[Ed. note: While we take some time to rest up over the holidays and prepare for next year, we are re-publishing our top ten posts for the year. Please enjoy our favorite work this year and we’ll see you in 2022.]
Remember the scene from the Matrix where Neo unlocks his full power, and the world around him is revealed as lines of code running in all directions? What if you could see the world around you in this way, so that the person sitting next to you was a webpage where one could right-click to inspect element and find the source code underneath?
We’re not quite there yet, but recent breakthroughs in nanopore sequencing, driven by developments in open-source software, have made it possible to greatly reduce the time it takes to decode a genome, shrinking what used to be a 15-day process to three days or less. It wasn’t so long ago that decoding a genome took years! To understand the code behind these new techniques, which have been dubbed UNCALLED, we chatted with Prof. Michael Schatz, the Bloomberg Distinguished Associate Professor of Computer Science and Biology at Johns Hopkins.
First, let’s start with a nanopore sequencer. “The idea for this originated about 30 years ago, and the legend is the first diagram was drawn on a napkin,” says Schatz. In reality the original concept for nanopore sequencing was sketched out by Dr. David Deamer (@UCSC_BSOE) in a stenographer's notebook using a red ink ballpoint pen!

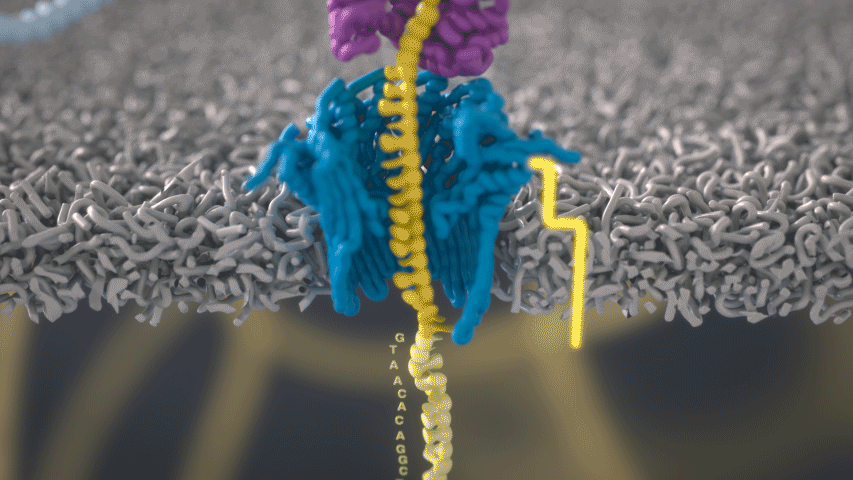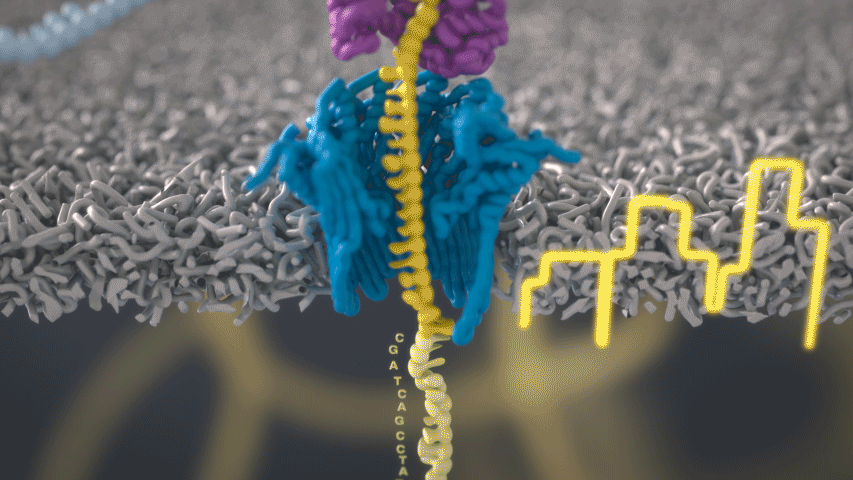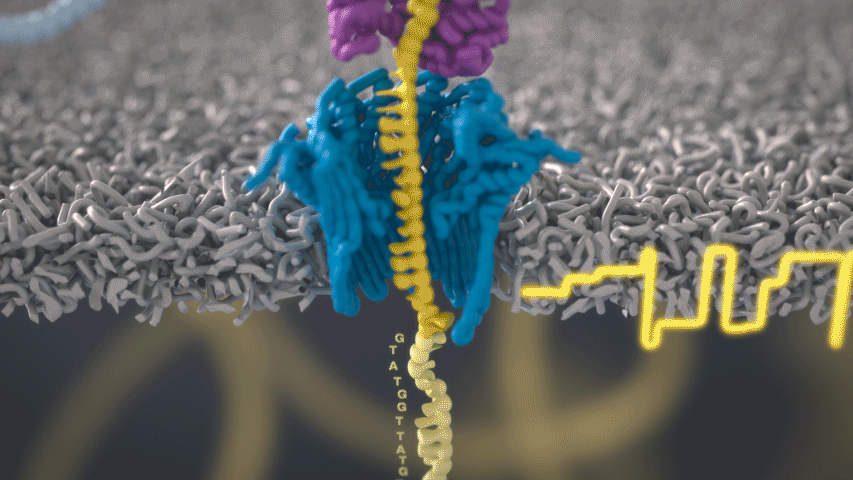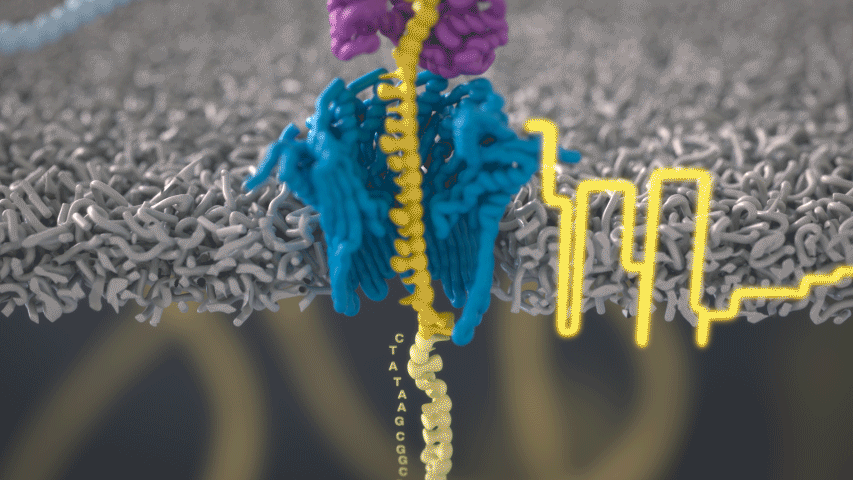
Imagine a hole so tiny that a single strand of DNA can fit through at a time. Push your genetic material through this pore, and the As, Ts, Gs, and Cs that make up a human genome will be revealed in sequence. So, how do you tell the four building blocks of DNA apart?
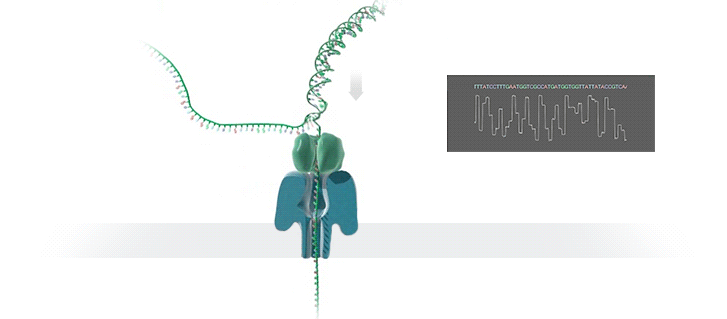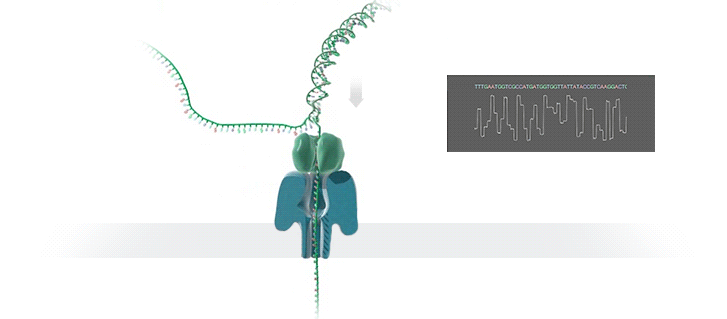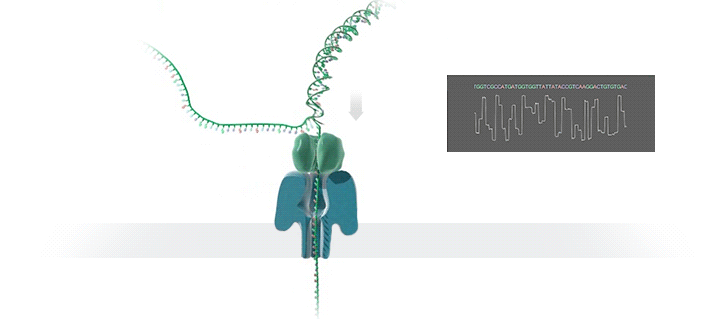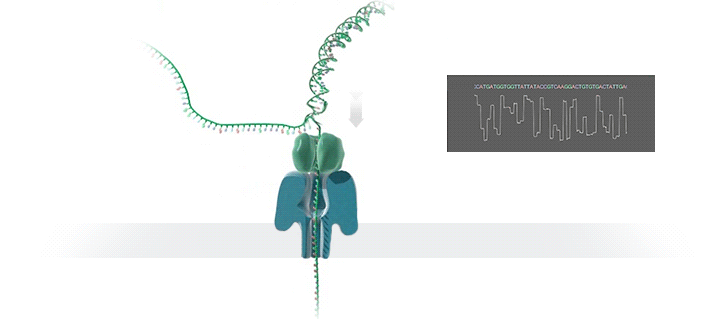
“It takes the most exquisite measurements you can imagine, measuring the changes in current associated with different bits of DNA,” he explains. “This is happening at the level of pico-amps—one-trillionth of an amp measurement—and we can get these readings in real time.” Five years ago, the equipment needed for this work would have been restricted to serious research facilities. Today, for about a thousand dollars, you can purchase a nanopore sequencer as a peripheral that connects to any computer via USB.
The sequencing produces very noisy electrical data, but Schatz and his team have developed a fuzzy logic inspired by a Markov model to decode each protein in near-real time. “I mean, it’s basically out of Star Trek, right?” says Schatz excitedly. “Nucleotides are passing through this tiny hole, and we’re measuring the current four thousand times a second.” The software is decoding the sequence in real time so that it can be matched to different genetic markers. So, for example, you could identify if it’s likely to be a pathogenic bacteria or a gene associated with cancer. More importantly, you can ignore fragments that aren’t of use at the moment.
Each bit of DNA passing through this little hole is a charged molecule. The software allows a user to actually reverse the voltage on an individual molecule, which has the effect of ejecting it out of the nanopore. It’s this ability to selectively sequence only the sections that are relevant to the work at hand which allows for such massive improvements in speed. “There is an API call to pick and choose which molecules you want to work with,” says Schatz. “It’s just amazing to me that this is even possible.”
Processing the language of life
Each DNA fragment returns a voltage reading based on its nucleotides. So, when you get a voltage, how hard is that lookup call? It’s not a simple table but rather some very fuzzy logic matching. “For the electrical data what you might want is for the A nucleotides, there's one particular current, for the C a different current, etc,” says Schatz. “But you don't get that at all.”
The electrical current is actually associated with several nucleotides in a row. About six nucleotides are the most influential. You can think of the electrical current like the DNA is being ratcheted through this little hole. “So you actually sense the same nucleotide about six different times in different contexts in six surrounding nucleotides.” The current is very noisy. For a particular current measurement there are typically hundreds of nucleotides sequences that it could possibly represent.
Think of each combination of those six having an offset. At offset one, there’s one hundred possible nucleotides sequences; at offset two, there's another hundred; at offset three, there's another hundred;, and at offset four, there's another hundred. “But it is in that combination of overlapping sequences that you can have any hopes to resolve this into a particular nucleotide since we know that the sequences must overlap.” For example, GATTACA at offset one could be followed by ATTACAT at offset two, but not TTTACAT, AATACAT, nor any other sequence that doesn’t begin ATTACA
The decoding uses a logic similar to natural language processing to match that noisy electrical signal to a nucleotide sequence.
Once you have the nucleotide sequence, you need to do text processing to decide where in the genome does this molecule originate from. “A lot of that technology was invented around database storage systems some 30 years ago,” says Schatz. “There's this really powerful data structure called the Burrows-Wheeler transform that is now really central to genomics these days.”
The nanopore sequencer is incredibly cheap compared to lab tools from a few years ago. But it does require a single use cartridge, called a flow cell, to sequence DNA molecules, and the cost of those can add up quickly when trying to look at large sequences. “What the software does is, rather than having to scan through the whole genome, we can be really picky about which molecules we're actually going to invest our sequencing into,” says Schatz. “We can pick and choose in real time which molecules will fully read out versus which molecules will eject after about one second of sequencing.”
So, for example, if you were looking to determine if a person was carrying a variant in a gene known to be associated with hereditary cancer, like BRCA1, you would take a sample. If you wanted to profile all the material with nanopore sequencing, that would be a pretty slow and expensive process. All the molecules are mixed up in a test tube and you sequence them one at a time as they're randomly pulled out of that collection. However, the new software from the Schatz lab called UNCALLED, led by Ph.D. student Sam Kovaka can evaluate in near real time if a sequence is worth studying or not.
In fact, during a normal sequence, you are likely to want to sequence the genome more than once, since any sample you take has a random collection of DNA molecules, and may not contain the parts you are most interested in. With the ability to select, you can winnow down what you’re looking for faster and avoid sequencing other areas over and over again.
Or, for example, take the example of infectious disease, which is on everyone’s mind these days. Labs around the world are struggling with massive workloads as testing explodes. “In that scenario, the human genome is kind of boring. That's not really what you're looking for.” Schatz says. With UNCALLED, the nanopore would eject anything obviously human. “Anything that doesn't match the human genome, we'll go back and we'll try to hold on to it and so we can do some real-time analysis of what it is.”
Open sourcing our source code
When Schatz first got into the world of genomics, the industry had a fairly bad reputation for being closed off and proprietary. “In the very early days, there was an effort to do a lot of gene patenting. There were some high profile cases about genes associated with breast cancer, for example. There were efforts to patent these sequences and charge extraordinary amounts of money to do what is now a very basic analysis.”
Luckily, says Schatz, that tendency has changed for the better in recent years. “There's been several waves of technologies over the last twenty years, so there's a real sense of urgency. Even though all those sequencers just write out the nucleotide sequences, every platform has different properties and characteristics and errors associated with it. So there's a real rush to develop software that can overcome these differences and make the best use of the data from the different platforms.”
Why not make the software into a proprietary product? Well, speed matters. “If you try to commercialize it, that takes a while to start a company, and it can take so long that by the time you go to the mechanics of that, the next thing has already emerged. There's such a race there that it's hard to commercialize the software for the long term.” Schatz continues, “Plus our work is largely funded through government sponsored grants, so this is one of the important ways for us to give back to society.”
The current climate is far healthier and happier for academics like Schatz, who plans to continue open sourcing the software being created by his lab. “There's just so much benefit from being able to share code and work collaboratively. In almost all cases the pros outweigh any sort of potential negatives.”
Illustrations by Alex Francis.